Computational Biology
Scribe Notes for Class 5
May 26, 2000
Scribe: L. Heath
Today's Handouts and Announcements
-
The class web site finally has the template for scribe notes,
so this week's scribes are requested to get their html
files in as quickly as possible,
preferably today.
Today's Topics
-
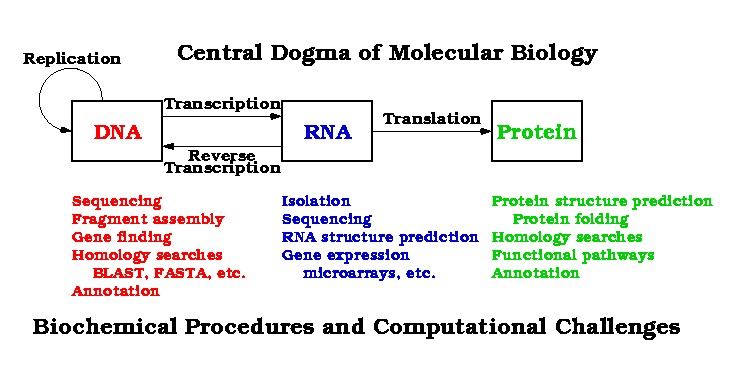
The Central Dogma of Molecular Biology
was discussed,
along with a set of computational challenges,
including these problems:
-
Fragment assembly ---
reconstructing large DNA sequences
by putting together many small contiguous sequences.
-
Gene finding ---
identify plausible open reading frames (ORFs)
within a large DNA sequence.
-
Homology search ---
take a DNA sequence
and search for similar sequences in sequence databases.
Uses algorithms such as BLAST and FASTA.
-
Annotation ---
use evidence from the literature,
experimentation, and homology searches
to attach functional information to a genetic sequence.
-
RNA structure prediction ---
predict 3-dimensional structure
from nucleotide sequence.
-
Gene expression ---
use information from microarray and other experiments
to identify genes expressed in particular cells under particular conditions.
Useful in understanding gene function.
-
Protein structure prediction ---
using the primary peptide sequence
of a protein and additional information from other sources,
determine how the protein folds
into its 3-dimensional shape.
Important for exploring protein function.
-
Homology search ---
take a peptide sequence
and search for similar peptide sequences in protein databases.
Uses algorithms such as BLAST and FASTA.
-
Annotation ---
use evidence from the literature,
experimentation, and homology searches
to attach functional information to a genetic sequence.
-
Functional pathways ---
identify functional relationships among proteins.
-
An introduction to
Genome Rearrangements
was made by John Paul Vergara.
-
Introduction ---
Mutations as genome rearrangements;
distances between genomes;
types of rearrangements: reversals, block moves, translocations
-
Complexity ---
Distance can be framed as a sorting problem
where a shortest sequence of permutations of a particular kind
is sought.
Computing the genome rearrangement distance
for most types of rearrangements seems very hard,
though there are polynomial-time solvable cases.
In particular,
minimum sorting by reversals is NP-hard (Caprara, 1999).
-
Minimum sorting by reversals ---
Kececioglu and Sankoff, 1995,
introduced the idea of breakpoints in a permutation.
They obtained a 2-approximation algorithm
for minimum sorting by reversals.
Subsequently,
Bafna and Pevzner, 1996,
introduced the idea of a breakpoint graph
and obtained a 3/2-approximation algorithm for signed permutations
and a 7/4-approximation algorithm for unsigned permutations.
Today's Sources
Please report any problems found in these pages to:
CS6104 Account (cs6104@courses.cs.vt.edu)
{kind=link}