Protein Science
(1998), 7:
1458-1468.
Cambridge University Press. Printed in the USA.
Copyright © 1998
The Protein Society
ARTICLE
Prediction of functional residues in
water channels and related proteins
A. FROGER,
1
B. TALLUR,
2
D. THOMAS,
1
and
C. DELAMARCHE
1
1 UPRES-A CNRS
6026, Biologie Cellulaire et Reproduction, Équipe
"Canaux et Récepteurs Membranaires,"
Université de Rennes1 bâtiment 13, Campus
de Beaulieu, 35042 Rennes Cedex, France
2 IRISA, Institut
de Recherche en Informatique et Systèmes Aléatoires,
Campus de Beaulieu, 35042 Rennes Cedex, France
(Received December 22, 1997;
Accepted February 25, 1998)
Reprint requests to:
Christian Delamarche, UPRES-A
CNRS 6026, Équipe Canaux et Récepteurs Membranaires
bâtiment 13, Campus de Beaulieu, 35042 Rennes Cedex,
France;
e-mail:cdelam@univ-rennes1.fr.
Abbreviations:
AQP, aquaporin protein;
CA, correspondence analysis;
GlpF, glycerol facilitator
protein;
MIP, major intrinsic
protein;
ORF, open reading frame;
PC, personal computer.
Abstract
In this paper, we present an updated classification
of the ubiquitous MIP (Major
Intrinsic Protein) family proteins,
including 153 fully or partially sequenced members available
in public databases. Presently, about 30 of these proteins
have been functionally characterized, exhibiting essentially
two distinct types of channel properties: (1) specific
water transport by the aquaporins, and (2) small neutral
solutes transport, such as glycerol by the glycerol facilitators.
Sequence alignments were used to predict amino acids and
motifs discriminant in channel specificity. The protein
sequences were also analyzed using statistical tools (comparisons
of means and correspondence analysis). Five key positions
were clearly identified where the residues are specific
for each functional subgroup and exhibit high dissimilar
physico-chemical properties. Moreover, we have found that
the putative channels for small neutral solutes clearly
differ from the aquaporins by the amino acid content and
the length of predicted loop regions, suggesting a substrate
filter function for these loops. From these results, we
propose a signature pattern for water transport.
Keywords:
aquaporin;
correspondence analysis;
glycerol facilitator;
MIP family;
multiple sequence alignment;
protein function prediction
Article Contents
(You can also go directly to the
beginning of the text.)
- Introduction
- Fig. 1. Predicted
membrane topology of the MIP family proteins
- Results
- The MIP family
- Table 1. Members
of the MIP family analyzed in this study
- Table 2. Sequences
of the MIP family analyzed to check the predictions
- Sequence alignment
analysis
- Fig. 2. Part
of the average similarity plots for the MIP family proteins
- Fig. 3. Portion
of the 40 multiple sequence alignment
- Conventional statistical
analysis
- Multivariate statistical
analysis
- Fig. 4. Correspondence
analysis applied on the MIP family proteins
- Discussion
- Equation 1
- Materials and methods
- Selection of protein
sequences
- Multiple alignment software
- Alignment analysis
- Correspondence analysis
- Acknowledgments
- References
Introduction
Water is the most ubiquitous molecule
of living systems and its movement across cell membranes
accompanies essential physiological functions. All biological
membranes exhibit some water permeability as a result of
diffusion through the lipid bilayer, under the driving
force of the osmotic gradient. However, some cells are
able to transport water at greatly accelerated rates by
way of water-selective channels called aquaporins. The
discovery of these specialized proteins has led to new
information on both the physiological and molecular mechanisms
of membrane water permeability and on links between aquaporins
and human diseases (King & Agre, 1996).
The first functionally characterized aquaporin, AQP1
(original name CHIP28), was discovered in the membrane
of red blood cells (Preston et al., 1992).
This protein is distributed in many water permeable tissues.
AQP1 is constitutively expressed in the proximal tubules
and descending thin limbs of the kidney where it mediates
90% of bulk water reabsorption (Sabolic & Brown,
1994). Presently, seven
other mammalian aquaporins have further been identified
(reviewed by Brown et al., 1995):
AQP2, the vasopressin sensitive water channel expressed
in the renal collecting tubules is implicated in a form
of nephrogenic diabetes insipidus; AQP3 also expressed
in kidney, exhibits water, glycerol and urea permeability;
AQP4 is predominantly expressed in the brain where it probably
plays a role in cerebrospinal fluid outflow regulation;
AQP5 is distributed in a variety of exocrine glands; AQP6
(hKID) is exclusively expressed in the kidney and is not
regulated by antidiuretic hormone. Finally AQP7 and AQP8,
two novel aquaporins, are predominantly expressed in testis
(Ishibashi et al., 1997a,
1997b), AQP7 being a
mixed channel, like AQP3.
Aquaporins have also been identified in amphibian,
plant, bacteria, and insect tissues. For example, FA-CHIP
has been characterized in frog urinary bladder (Abrami
et al., 1994), gamma-TIP
and RD28 have been identified, respectively, in the tonoplast
and the plasma membrane of Arabidopsis thaliana
(Maurel et al., 1993;
Daniels et al., 1994).
AqpZ was discovered in Escherichia coli
where it seems to play a role in osmoadaptation by contributing
to cellular volume regulation in hypoosmolar media (Calamita
et al., 1995, 1997). One aquaporin, called AQPcic,
was recently characterized in the digestive tract of an
homopteran sap-sucking insect, Cicadella viridis
(Beuron et al., 1995;
Le Cahérec et al., 1996).
The aquaporins belong to an ancient and ubiquitous
family of channel proteins called the MIP
family with reference to MIP26 (AQP0), the Major
Intrinsic Protein expressed in lens fiber cells
(Gorin et al., 1984).
The function of the archetype MIP26 is unclear. When expressed
in Xenopus oocytes, MIP26 weakly enhances permeability
for ions, water, and glycerol, suggesting multiple physiological
functions for this protein (Chandy et al., 1995; Kushmerick et al., 1995; Mulders et al., 1995).
Sequence comparisons revealed that the glycerol facilitators
(GlpF) are also members of the MIP
family. Bacteria use glycerol as a carbon source for glycolysis,
and for lipid biogenesis. The transport of glycerol into
the cytoplasm involves two types of mechanisms: a passive
diffusion across the lipid bilayer, or a facilitated uptake
when the external concentration of glycerol is low (Richey
& Lin, 1972). The
GlpF protein acts as a selective pore
for uncharged molecules, depending on the molecular size
of the substrates (Heller et al., 1980;
Sanders et al., 1997).
A glpF gene has been cloned from Bacillus
subtilis, E. coli, Haemophilus
influenzae, Pseudomonas aeruginosa,
and Shigella flexneri. A glycerol
facilitator was also characterized in Saccharomyces
cerevisiae (Luyten et al., 1995).
Members of the MIP family are now
included in the PROSITE database (Bairoch et al., 1996) with the signature sequence
[HNQA]-x-N-P-[STA]-[LIVMF]-[ST]-[LIVMF]-[GSTAFY], with
x as any residue and alternative residues in brackets.
All the MIP proteins are about 260 residues
long, with the exception for two MIP yeast
proteins that have more than 600 residues, as a result
of extended N- and C-terminal segments. Hydropathy plots
and experimental investigations by epitope insertions and
mutagenesis reveal a common topology for these molecules,
with six transmembrane domains. After sequence alignment,
the most divergent members of the family display less than
20% identity, but like the other MIP
proteins, they share highly conserved residues distributed
throughout the sequence. A characteristic homology common
to the members of the MIP family is the
Asn-Pro-Ala (NPA) motif repeated in two opposite loops
(Fig. 1). Moreover,
the NH2 and COOH-terminal halves are sequence-related,
suggesting an ancestral intragenic gene duplication as
known for other channel families (Pao et al., 1991; Wistow et al., 1991; Reizer et al., 1993; Saier, 1994).
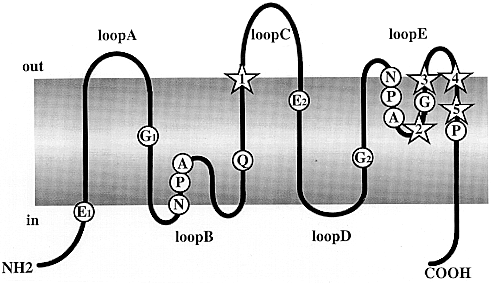
Fig. 1
Predicted membrane topology of the MIP
family proteins. Primary structure of
a monomer based on the hourglass model of aquaporins proposed
by Jung et al. (1994).
The molecule consists of six membrane-spanning domains
connected by loops A to E, with cytoplasmic NH2- and COOH-
termini. Highly conserved residues including the two Asn-Pro-Ala
(NPA) repeats are indicated. The stars indicate positions
1 to 5 predicted from the present study to play a functional
role. In the hourglass model, loops B and E protrude into
the lipid bilayer, and the NPA boxes joints themselves
in the middle of the channel to form a single aqueous pathway.
Indices are used to identify segments studied in the correspondence
analysis.
The detection of a functional and/or structural site
in a nucleic acid or protein is of major interest. Unfortunately,
there is no simple and straightforward method, since the
specific properties of a given molecule often result from
a limited number of key residues. For example, single nucleotide
changes in given positions of tRNATrp and tRNATyr
are sufficient to transform the identity of these tRNAs
to glutamine (Cavarelli & Moras, 1993).
Single amino acid substitutions also modify the ion-selection
properties of the sodium channel protein into a calcium
channel (Heinemann et al., 1992).
The most common way to find the residues of functional
importance is by site-directed mutagenesis. However, to
avoid random targeting that can be both laborious and expensive,
computational methods are of particular relevance. Since
the enigmas of the structure/function of proteins are hidden
within their sequences, a careful comparison of homologous
sequences from a large number of organisms should enable
us to make certain predictions.
Functionally, for the MIP family,
we know that the aquaporins present a highly selective,
but quantitatively heterogenous, water permeability (Yang
& Verkman, 1997),
and that the GlpF channels transport glycerol
but exclude water. Therefore, in order to understand the
molecular mechanisms of substrate selectivity and permeation,
one must find the residues that are specifically linked
to each subclass. In this report, we present the results
obtained from a sequence analysis of the MIP
proteins and point out some residues that could modulate
the selectivity for water or glycerol. Two different approaches
were used: (1) a systematic comparison of the physico-chemical
properties of the amino acids at each position in multiple
sequence alignments and (2) a statistical analysis (conventional
and multivariate) to compare the amino acid composition
in sequence segments. The results are assessed by comparison
with published experimental data.
Results
The MIP family
In Tables
1 and 2,
we present an updated compilation of 122 complete and 27
partial sequences of the MIP family available
from public databases.
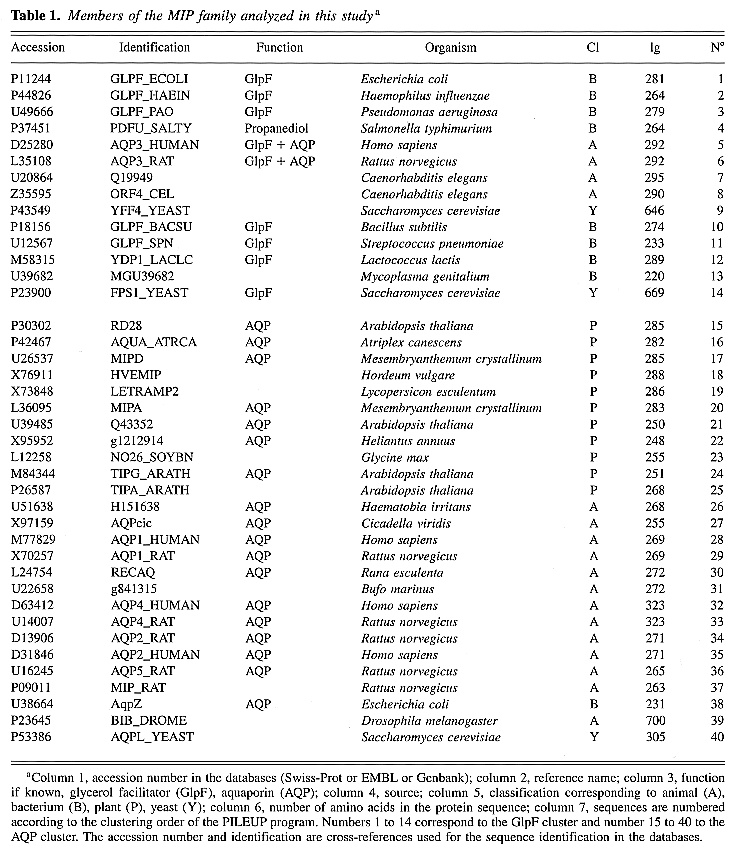
Table 1. Members of the MIP
family analyzed in this studya
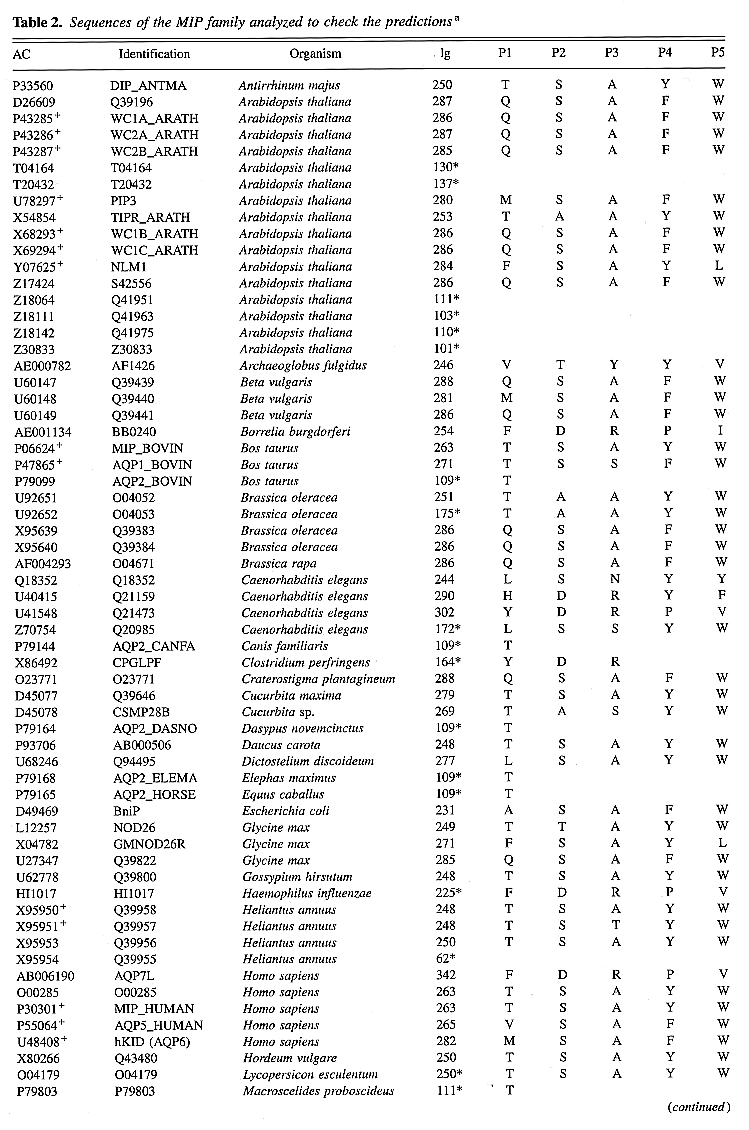
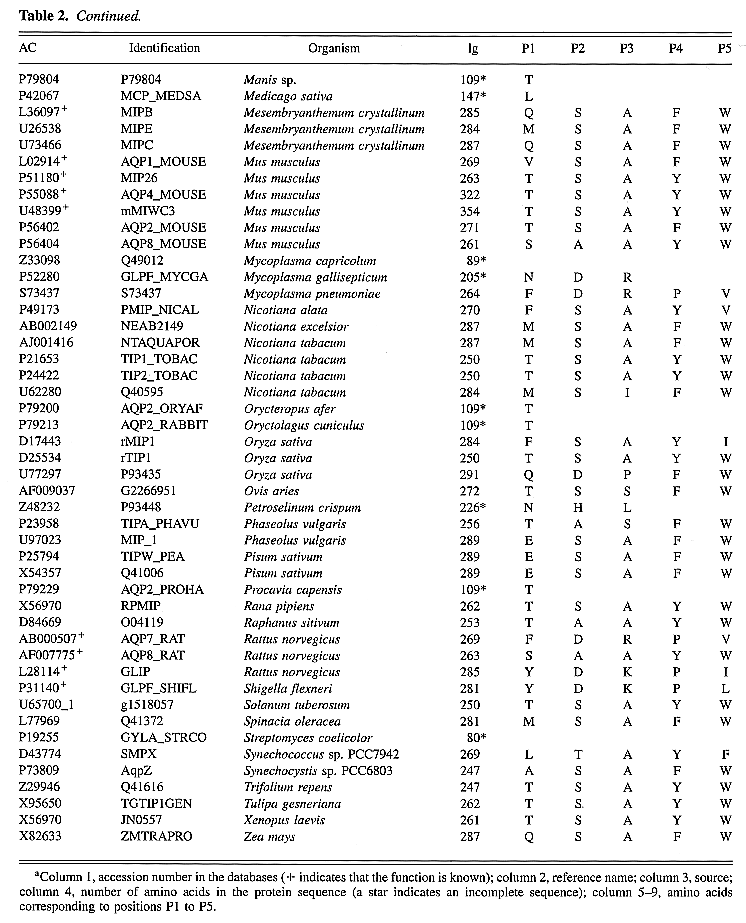
Table 2. Sequences of the MIP
family analyzed to check the predictionsa
These sequences were retrieved by combining searches with
specific keywords and/or with the PROSITE signature sequence
for the MIP family. The number of MIP
sequences has rapidly increased since the recent publication
of 84 members by Park and Saier (1996).
However, the last update of the PROSITE database (PS00221,
November 1997) lists only 63 MIP proteins.
All of the proteins presented in Tables
1 and 2
have a nonredundant accession number in the databases.
It is obvious that only a small number of closed sequences
results from genetic polymorphism. For example, AqpZ (U38664)
differs from Bnip (D49469) by only three amino acids, but
both are homologous genes of different E. coli
strains.
Proteins of the MIP family are abundant
in plant cells, where they are expressed both in plasma
and vascular membranes. For A. thaliana,
the model plant for genome sequencing, we retrieved 20
different amino acid sequences from standard databases,
but found 84 sequences (not shown) in the specialized database
of the Institute for Genomic Research (http://www.tigr.org). Most of A.
thaliana MIPs are believed to be aquaporins.
The plant aquaporins play important physiological roles,
including different levels of water permeability, probably
for adaptation to a variety of water stresses and in relation
to the plant cell compartmentalization (Maurel et al.,
1997; Weig et al., 1997).
Complete genome sequences provide the opportunity
to examine the presence of MIP sequences
in these genomes. Twelve complete genomes were examined:
S. cerevisiae possess four MIP
encoding genes, one of them, Q12302, cited in the conclusion
paragraph, contains a frameshift mutation. E.
coli possess two distinct and functionally identified
MIP proteins, one aquaporin and one glycerol
facilitator. Two distinct MIP coding sequences
have also been described in the genome of H.
influenzae but only one in that of Archaeoglobus
fulgidus, Bacillus subtilis, Borrelia burgdorferi, Mycoplasma
genitalium, Mycoplasma pneumoniae,
and Synechocystis sp. Interestingly,
no MIP-protein-encoding sequences were
retrieved from the genomes of Helicobacter pylori
(eubacteria), Methanobacterium thermoautotrophicum,
and Methanococcus jannaschii (archeobacteria).
Sequence alignment analysis
The quality of the alignments obtained
using various software was similar. The major difference
was in the number of gaps introduced into the alignments.
For this reason, further experiments were done with two
independent methods: a precise analysis of the alignment
content in regions of high conservation between the sequences,
and a statistical analysis on sequence segments less dependent
on alignment methods.
The PILEUP program was used to
create a multiple alignment with the test set of 40 sequences.
In the resulting dendrogram, the data are clearly separated
into two major clusters and each cluster fits into a main
functional subgroup: cluster I corresponds to glycerol
transport and cluster II corresponds to water transport
(Table 1). In all the
sequence alignment studies, the proteins AQP3 (Nos. 5 and
6), which are able to transport either glycerol or water
when expressed in the Xenopus oocyte,
were allocated to cluster I. MIP26, the archetype of the
MIP family, was allocated to cluster II.
The function of some of the 40 proteins is yet unknown,
but the partition into two major clusters was not affected
by adding or removing new sequences to create the multiple
alignment. This is in agreement with a recent phylogenetic
study (Park & Saier, 1996),
concluding that most and probably all the MIP
family members will exhibit specificity for water and/or
small neutral solutes. At this point in our analysis, we
postulated that it should be possible to identify residues
that are directly linked to function by a careful inspection
of the multiple alignments. In the following experiments,
we will consider the alignment of 40 sequences and the
two subgroups extracted from this alignment: cluster I,
14 sequences, which will be named the GlpF
cluster, and cluster II, 26 sequences, which will be named
the AQP cluster.
One of the most popular prediction methods for getting
information concerning the protein membrane topology consists
in averaging the residue hydropathy for consecutive 19-residue
segments (Kyte & Doolittle, 1982).
We compared the average hydrophobicity profiles of the
two clusters, using different hydrophobicity scales (data
not shown). The resulting profile supports the prediction
of six membrane spanning segments for the MIP
proteins (Reizer et al., 1993).
The transmembrane segments are clearly superimposable for
the two functional subgroups, while for predicted loop
segments, the hydrophobicity scores differ significantly,
suggesting the presence of residues linked to function.
The hydrophobicity profile method is based on an average
value calculated along a sliding window. Thus, the presence
of gaps in the alignment will alter the comparison between
the two clusters. Therefore, we used another method to
extract the information at each position of the alignment:
the similarity profile method, which was applied with a
one residue window (Fig. 2).
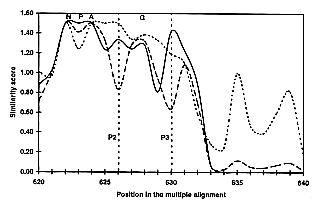
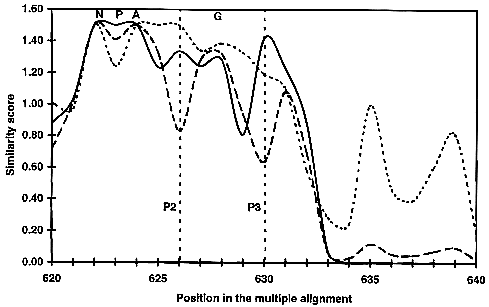
Fig. 2
Part of the average similarity plots for
the MIP family proteins. At each position
of the 40 sequence alignment, the average similarity score
is calculated for each cluster. The similarity score was
calculated using the modified Dayhoff table (Gribskov &
Burgess, 1986) scale,
and plotted with a sliding window of one. This figure illustrates
the similarity plot from position 620 to 640 of the global
alignment. The similarity for the 40 sequences is shown
as a broken curve, the GlpF cluster is
shown as a dotted curve and the AQP cluster
as a continuous curve. A maximum score of 1.5 is obtained
when all amino acids at a given position of the alignment
are identical. Highly conserved residues presented in Figure 1 are indicated above the alignment.
The positions P2 and P3 predicted to have a functional
role are indicated.
Potential residues of high interest are directly visualized
on the curves, when the similarity score for each cluster
is higher than the score for all sequences. Five discriminating
positions were identified, where the physicochemical properties
are conserved within each subgroup, but differ between
the subgroups. These positions are located in highly conserved
regions, and can be easily retrieved from any sequence
(Figs. 1, 3).

Fig. 3
Portion of the 40 multiple sequence alignment.
Sequences are numbered according to Table
1 and the position of the first residue in
each sequence segment is indicated in parenthesis. The
positions P1 to P5 predicted to have a functional role
in the MIP proteins are boxed. The highly
conserved residues presented in Figure
1 are indicated below the alignment (*).
From our observations, we deduced the following rule:
Position 1, located in the terminal part of the third
transmembrane segment, is an aromatic residue in the GlpF
cluster. This residue is not aromatic in the AQP
cluster.
Positions 2 and 3 are located in loop E, just behind
the second "NPA" box. They correspond respectively
to an acidic then a basic residue (D, then R or K) in the
GlpF cluster and to two small uncharged
residues in the AQP cluster.
Positions 4 and 5 correspond to two consecutive amino
acids located in the sixth transmembrane segment. These
positions can be defined as two aromatic residues in the
AQP cluster compared with a proline followed
by a nonaromatic residue in the GlpF cluster
(except for the yeast protein YFF4, which possess an alanine
in P4 and a tryptophan in P5).
A close inspection of the second set of sequences
(Table 2) confirms that
the five positions described above are composed of remarkably
conserved residues. Of 112 proteins, 21 are functionally
characterized, and 20 of them present a perfect correspondence
with the rule, confirming possible functional role for
key residues. The exception concerns a newly characterized
aquaporin from A. thaliana, NLM1 (Weig
et al., 1997), which
possess mixed key residues of GlpF cluster
for P1 and P5, and AQP cluster for P2-P4.
Of 450 key residues available from Table
2, only 22 (4.8%) differ from those
observed in Figure 3.
In the absence of a functional characterization of the
corresponding proteins, it is difficult to relate these
differences to extensions or divergences of the rule, or
to errors in the sequences. Residues particularly well
conserved concern each of the couples P2-P3 and P4-P5.
These couples are closely tied to each other: charges of
opposite sign in P2-P3 associate with nonaromatic
residues in P4-P5 or uncharged residues in P2-P3
associate with aromatic residues in P4-P5. Presently,
there are only two exceptions to this observation and they
concern two MIP proteins of Caenorhabditis
elegans (U40415, Z35595). Another observation
concerns the position P1, which is generally the counterpart
of P4-P5 (aromatic/nonaromatic) between the two clusters.
Conventional statistical
analysis
The differences between the glycerol facilitators
and the aquaporins, revealed by the average similarity
profiles, result in changes of amino acid content between
the subgroups. These compositional differences can be quantified
by conventional statistical analysis. The mean amino acid
content and the mean length in amino acids of the segments,
predicted to be inside or outside the membrane, were calculated
for each subgroup and compared with the "Student
t" test. Such a trivial analysis
revealed some of the residues that participate in the key
positions: position 1 in TM3 (Y), position 2 and 3 in loop
E (D,R) and position 4 in TM6 (P), as defined previously.
We also observed that some amino acids are over-represented
in glycerol facilitators (W in TM2; P in loop B; I, F,
P, T in loop C; N in TM4 and loop D; G, L in loop E), and
that others are significantly more frequent in aquaporins
(A, S, W in TM1; V in loop B; C in TM3, R and K in loop
D; H in TM5 and TM6). Moreover, the statistical analysis
revealed significant differences (Student t
test, P < 0.001) in the predicted
length of two external loops. Loops C and E are longer
for the GlpF cluster than for the AQP
cluster. The respective mean lengths, calculated from the
40 sequences of Table 1
are 27.92 compared with 18.24 for loop C and 28.35 compared
with 18.52 for loop E.
The results of the above comparisons strongly suggest
that several amino acids could contribute to structural
and functional differences between the glycerol facilitators
and the aquaporins. However, these results were obtained
by averaging the amino acid content over already predetermined
functional subgroups. Thus, to check our observations without
a priori classification, we have carried out a multivariate
analysis on the 40 MIP proteins.
Multivariate statistical
analysis
Correspondence analysis was used to compare
the amino acid frequencies in the MIP
proteins, from segments of different lengths. CA
does not take into account the amino acid order along the
polypeptide chain or the presence of gaps in the alignment.
The results of the correspondence analysis are shown on
a factorial map in which each amino acid is represented
by its letter, and each MIP protein by
the number corresponding to the sequence in Table 1. Proteins having a close resemblance
are positioned closely on the map. Thus, if the two functional
subgroups are clearly separated on the map, this suggests
that the segment participates in the channel specificity.
Preliminary experiments using entire sequences or short
segments gave unsatisfactory results. The best separations
were obtained for medium-sized segments located between
but excluding two strictly conserved amino acids. These
selected segments are marked on Figure
1 from NH2- to COOH termini: E1-G1,
G1-Q, Q-E2, E2-G2,
G2-P. In our study, the projection map obtained
with the segment Q-E2, including the major part
of the third transmembrane domain and loop C, gave a clear
and complete separation of the two functional subgroups
(Fig. 4).
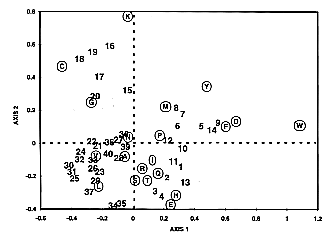
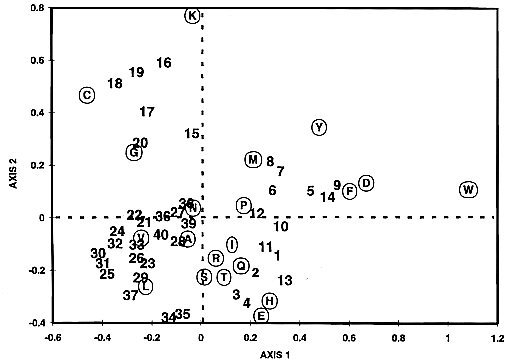
Fig. 4
Correspondence analysis applied on the
MIP family proteins. Correspondence analysis
was applied to the segment located between the residues
Q and E2, principally including the third transmembrane
domain and loop C (Fig. 1).
Each segment in the MIP proteins is represented
as a vector point in a 20-dimensional space, where each
dimension corresponds to the relative frequency of one
of the 20 amino acids. The cloud of proteins as well as
that of amino acids are then projected on the plane of
first and second factors and these projections are overlaid.
Proteins of similar composition appear as neighbors and
amino acids contributing to the separation are easily identified.
The first factor accounts for 20.9% of the total
variance between MIP proteins, while factors
2, 3, and 4 account for 13.7%, 10.4%, and
9.7%, respectively. Thus, the first factor corresponds
to the most predominant differences present in the population
sequence. Along this factor (F1), all glycerol facilitators
have positive values and all aquaporins have negative ones.
The simultaneous representation of proteins and amino acids
leads to sensitive means of identifying those amino acids
responsible for the separation on the factor map. Tryptophane,
aspartic acid, phenylalanine, and tyrosine have a high
relative contribution on the first factor to separate glycerol
facilitators and aquaporins. These results support our
key residues rule given for position P1 (located inside
the Q-E2 segment): aromatic residues (phenylalanine
and tyrosine) are characteristic of glycerol facilitators.
Along factor 2, aquaporins are separated into two major
subgroups, one of which is a subgroup of plant aquaporins
(numbers 15 to 20). Lysine, cysteine, and glycine seem
responsible for this separation. Intriguingly, two AQP2
aquaporins, from rat and human (numbers 34 and 35), are
positioned in the extreme negative values of factor 2.
The presence of glutamic acid in their segment appears
to be responsible for this separation.
Discussion
Recently, Park and Saier (1996) reported a phylogenic study
of the MIP family based on the comparison
of 84 protein sequences. The authors concluded that most,
if not all, MIP proteins fall into one
of two physiological groups: (1) the water transport by
the aquaporins and (2) the small neutral solutes transport
such as glycerol by the glycerol facilitators. Considering
these two main functions, the present work focuses on the
characterization of functional residues in the MIP
proteins. The study was carried out by the analysis of
multiple protein sequence alignments, a standard strategy
to highlight residues of structural and functional importance
in a protein family (Livingstone & Barton, 1993), and by a new strategy based
on amino acid frequencies in sequence segments.
The present work highlights five key positions that
could play an important role in the structure and function
of each MIP family subgroup, and consequently,
raises questions on the mechanisms of substrate selectivity
by conserved residues. An hourglass topological model was
previously proposed for AQP1 (Jung et al., 1994). In this model, the loops B
and E dip and join into the channel. According to this
scheme, the second "NPA" region, which includes
P2 and P3, should be very close to P4 and P5 and possibly
to P1 (Fig. 1), thus
forming an atomic network whose bond properties are differentiated
by these key residues, resulting in channel specificity.
However, as we have also shown, the amino acid length and
composition of the external loops C and E differ significantly
from glycerol facilitators to aquaporins, and these parameters
may probably play a crucial role in pore selectivity. In
summary, we predict that the external aperture of the channel,
surrounded by the key residues and protected by the external
loops act as a substrate filter. Further sequence analysis
will be required to combine our observations in a single
rule, and to identify the amino acids able to interact
at distance. Mutagenesis experiments on AQPcic (an insect
aquaporin) and on GlpF (the glycerol facilitator
of E. coli) are currently in progress
in our laboratory to assess these predictions. Problems
with this experimental approach is that missense mutations
can induce multiple effects, particularly in protein folding
and trafficking (Mulders et al., 1996).
In these conditions, it is difficult to determine if the
mutation also affects the functional properties of the
channel.
Meanwhile, by examining the current literature in
the field, we can evaluate the accuracy of our predictions.
The following results support our predictions:
The insertion of 25 amino acids in loop
E of hAQP1 (V201-E1) resulted in a markedly reduced water
channel activity (Preston et al., 1994).
We have shown by statistical analysis that the length of
this loop is significantly longer in glycerol facilitators
than in aquaporins.
In the aquaporin hAQP2, the substitution
of a segment of loop C (eight amino acids) by an other
segment of loop C (eight amino acids) from GlpF
abolished water transport function (Bai et al., 1996). This substitution introduced
into the loop two proline, one isoleucine, and one phenylalanine,
residues that we demonstrated to be more abundant in loop
C in glycerol facilitator than in aquaporins.
The substitution of a serine by a cysteine
at position 196 of hAQP1 greatly reduced the water transport
activity of this aquaporin (Jung et al., 1994).
This serine corresponds to position P2 in our rule.
The aquaporin AQP7 was recently cloned from
rat testis. Expression experiments in Xenopus
oocytes have shown that AQP7 also transports glycerol (Ishibashi
et al., 1997a, 1997b). The five key residues of
AQP7 are Y, D, R, P, and V and correspond, according to
our rule, to the solute transport signature. Interestingly,
like AQP7, AQP3, which is also a mixed-channel, bears the
signature of glycerol facilitators, as if the key residues
are more important for solute transport than for water
transport.
The results presented below do not follow our predictions
but allows refinements of our rule:
The insertion of 25 amino acids
in loop C of hAQP1 (T120-E1) had no significant effect
on water permeability (Preston et al., 1994),
even though this loop is significantly longer in glycerol
facilitators. This result suggests that the length of loop
C does not affect the water permeability but probably the
solute transport properties.
The permeability to water and glycerol for
MIP26 and NOD26 have been recently demonstrated (Kushmerick
et al., 1995; Mulders
et al., 1995; Rivers
et al., 1997). These
functional properties should assign the two proteins to
cluster I, by analogy with AQP3. However, MIP26 and NOD26
also exhibit ion channel activity (Weaver et al., 1994; Kushmerick et al., 1995; Lee et al., 1995;
Modesto et al., 1996).
The SmpX protein (D43774) of the Gram-negative bacterium
Synechococcus might also be involved
in ion transport, particularly in copper transport (Kashiwagi
et al., 1995). Therefore,
cluster II proteins could include functional subgroups
other than water transport.
A great number of published MIP sequences
were obtained by the RT-PCR technique using degenerate
primers directed against the two conserved "NPA"
boxes. Moreover, a large number of sequences were obtained
from poly(A)+ RNA prepared from the kidneys
of vertebrates, and in plants, most MIP
genes were cloned after induction by water-deficit stress.
As a result of such strategies, a bias could be expected
with a great number of published MIP sequences
being aquaporins bearing the two "NPA" boxes.
A question arises immediately: Is it possible to define
aquaporin sequences without reference to the "NPA"
boxes?
We examined this question by retrieving all sequences
in the databases matching with a new signature sequence,
which excludes the "NPA" motif. We have focused
this preliminary search on key amino acids P2 to P5 deduced
from the 26 sequences of cluster II (Fig.
3), and located between highly conserved
residues (G2-P, Fig. 1).
From these, we designed a signature pattern specific for
water but not for solute transport:
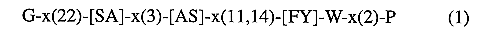
(the signature is reported in the PROSITE format, x corresponds
to any residue and brackets to alternative residues).
This signature sequence based on only six residues
was used to extract related proteins from the Swiss-Prot
and the TREMBL databases. These databases include more
than 170,000 entries corresponding to the translation of
all CDS in the EMBL Nucleotide Sequence Database (Apweiler
et al., 1997). We retrieved
91 sequences, from which 81 sequences have, as expected,
the signature we proposed for aquaporin and possess the
NPA boxes. From these 81 sequences, we have retrieved four
new MIP sequence proteins: U58207 from
Allium cepa, P28238 from Gallus
gallus, Q12302 from S. cerevisiae
and P93683 from Sorghum bicolor. These
sequences were not retrieved in our previous analysis because
they correspond to partial sequences without the first
"NPA" box. Consequently, the total number of
MIP sequences recorded in this study is
153. The other ten sequences retained with the signature
sequence do not appear to be, at a first glance, water
channels (P80517, Q09652, P44843, O00213, P30208, P47542,
P78018, Q10782, P46933, P74003). In conclusion, we have
shown that, if the presence of the "NPA" boxes
is a characteristic of the MIP family,
water transport function can be described by using a specific
signature devoid of any "NPA" box, such as
AQP6 (AB006190).
On the basis of the constantly increasing number of
sequences available for the MIP family,
together with more functional characterizations, we believe
that a similar approach is feasible to design a specific
signature for other transport functions.
Materials and methods
Selection of protein sequences
The sequences were extracted from GenBank,
EMBL, and PROSITE databases (indexing date Dec 1997). We
based our initial analysis on a test set of 40 MIP
protein sequences representative of different groups of
organisms (Table 1).
In order to avoid any bias in the analysis, we took care
to include divergent members rather than a too large number
of very similar ones. This initial analysis was limited
by the number of available sequences. Therefore, another
set, including 109 other fully or partially sequenced MIP
proteins, was used to amplify the predictive reliability
of the results (Table 2).
Multiple alignment software
We have used three multiple alignment
programs, using various values for the critical parameters,
K-tuple and gap penalties: CLUSTALW
(Thompson et al., 1994),
MAP (Huang, 1994),
and PILEUP from the GCG
package (Devereux et al., 1984).
We have tried three amino acid substitution tables: the
Dayhoff table PAM250, (Dayhoff et al., 1978),
the standard GCG table (Gribskov &
Burgess, 1986), and
the BLOSUM62 table (Henikoff & Henikoff, 1992). The computation from the GCG
programs was performed using INFOBIOGEN resources (http://www.infobiogen.fr).
Alignment analysis
Assignments for transmembrane domains
were made with the TMAP program (Persson
& Argos, 1994),
available on the Worldwide Web (http://www.embl-heidelberg.de/tmap/tmap_mul.html).
The program CGD (Delamarche, unpubl.
obs.) was used to analyze the amino acid composition along
the sequence alignments: amino acid frequencies, similarities,
charge distribution, hydrophobicity, etc. CGD
runs on PC using the powerful computing
and graphical tools of EXCEL worksheets.
For the hydrophobicity profiles, each of the three hydropathy
scales was used: Hopp and Woods (1981),
Kyte and Doolittle (1982),
Rao and Argos (1986).
For the similarity profiles, CGD uses
the same substitution matrices than for the multiple alignments.
The similarity score at a given position of the alignment
is the arithmetic mean of all the pairwise amino acid similarity
scores at that position. The distance between the couples
formed by two gaps or one gap and one amino acid is zero.
In this way, a position in the alignment that contains
many gaps will have a lower similarity score. This algorithm
is similar to that used by the program PLOTSIMILARITY
from the GCG package (Devereux et
al., 1984).
Correspondence analysis
Correspondence analysis (CA)
(Benzecri, 1973) may
be applied to contingency tables whose rows and columns
correspond, respectively, to "individuals"
(or objects) and attributes (or categories). In our study,
the data consist of 40 * 20 contingency tables whose
rows correspond to 40 MIP protein sequences
(entire sequences or segments situated between two strictly
conserved amino acids) and whose columns represent 20 amino
acids. The (i,j)
cell of the contingency table contains the frequency of
amino acid j in sequence i.
CA was carried out on entire sequences
and on five different segments using the CORRESP procedure
of SAS/STAT software. In this paper,
we present the results obtained on the segment Q-E2, which
gave the highest inertia.
Acknowledgments
The authors thank Dr. Rebecca
Hartley and Dr Isabelle Pellerin for helpful discussions.
This work was supported by the Langlois Foundation
(Rennes, France).
Abrami L, Simon M, Rousselet
G, Berthonaud V, Buhler JM, Ripoche P. 1994.
Sequence and functional expression of an amphibian
water channel: A new member of the MIP family.
Biochem Biophys Acta 1192:147-151.
Apweiler R, Gateau A, Contrino
S, Martin MJ, Junker V, O'Donovan C, Lang F, Mitaritonna
N, Kappus S, Bairoch A. 1997.
Protein sequence annotation in the genome era:
The annotation concept of SWISS-PROT+TREMBL.
ISMB 5:33-43.
Bai L, Fushimi K, Sasaki
S, Marumo F. 1996. Structure
of aquaporin-2 vasopressin water channel. J
Biol Chem 271:5171-5176.
Bairoch A, Bucher P, Hofmann
K. 1996. The PROSITE
database, its status in 1995. Nucleic
Acids Res 24:189-196.
Benzecri JP.
1973. L'Analyse des Correspondances,
tome2. Paris: Dunod.
Beuron F, Le Cahérec
F, Guillam G, Cavalier A, Garret A, Tassan JP, Delamarche
C, Schultz P, Mallouh V, Rolland JP, Hubert JFH, Gouranton
J, Thomas D. 1995. Structural
analysis of a MIP family protein from the digestive tract
of Cicadella viridis.
J Biol Chem 270:17414-17422.
Brown D, Katsura T, Kawashima
M, Verkman AS, Sabolic I. 1995.
Cellular distribution of the aquaporins: A family
of water channel proteins. Histochem
Cell Biol 104:1-9.
Calamita G, Bishai WR,
Preston GM, Guggino WB, Agre P. 1995.
Molecular cloning and characterization of AQP2,
a water channel from Escherichia coli.
J Biol Chem 270:29063-29066.
Calamita G, Kempf B, Rudd
KE, Bonhivers M, Kneip S, Bishai W, Bremer E, Agre P.
1997. The aquaporin-Z water
channel gene of Escherichia coli:
Structure, organization and phylogeny. Biol
Cell 89:321-329.
Cavarelli J, Moras D.
1993. Recognition of tRNAs
by aminoacyl-tRNA synthetases. FASEB
J 7:79-86.
Chandy G, Kreman M, Laidlaw
DL, Zampighi GA, Hall JE. 1995.
The water permeability per molecule of MIP is
less than that of CHIP. Biophys J
68A:353.
Daniels MJ, Mirkov TE,
Chrispeels MJ. 1994. The
plasma membrane of Arabidopsis thaliana
contains a mercury-insensitive aquaporin that is a homolog
of the tonoplast water channel protein TIP.
Plant Physiol 106:1325-1333.
Dayhoff MO, Schwartz
RM, Orcutt BC. 1978. A
model of evolutionary changes in proteins.
In Dayhoff MO, ed. Atlas
of protein sequence and structure, vol 5, suppl 3.
Washington, DC: National Biochemical
Research Foundation. pp 345-358.
Devereux J, Haeberli P,
Smithies O. 1984. A
comprehensive set of sequence analysis programs for the
VAX. Nucleic Acids Res 12:387-395.
Gorin MB, Yancey SB, Cline
J, Revel JP, Horwitz J. 1984.
The major intrinsic protein (MIP) of the bovine
lens fiber membrane: Characterization and structure based
on cDNA cloning. Cell 39:49-59.
Gribskov M, Burgess RR.
1986. Sigma factors from
E. coli, B. subtilis,
phage SPO1, and phage T4 are homologous proteins.
Nucleic Acids Res 14:6745-6763.
Heinemann SH, Terlau H,
Stuhmer W, Imoto K, Numa S. 1992.
Calcium channel characteristics conferred on
the sodium channel by single mutations. Nature
356:441-443.
Heller KB, Lin ECC, Wilson
TH. 1980. Substrate
specificity and transport properties of the glycerol facilitator
of Escherichia coli. J
Bacteriol 144:274-278.
Henikoff S, Henikoff GJ.
1992. Amino acid substitution
matrices from protein blocks. Proc
Natl Acad Sci USA 89:10915-10919.
Hopp TP, Woods KR.
1981. Prediction of protein
antigenic determinants from amino acid sequences.
Proc Natl Acad Sci USA 78:3824-3828.
Huang X. 1994.
On global sequence alignment. Comput
Applic Biosci 10:227-235.
Ishibashi K, Kuwahara
M, Gu Y, Kageyama Y, Tohsaka A, Suzuki F, Marumo F, Sasaki
S. 1997a. Cloning
and functional expression of a new water channel abundantly
expressed in the testis permeable to water, glycerol, and
urea. J Biol Chem 272:20782-20786.
Ishibashi K, Kuwahara
M, Kageyama Y, Tohsaka A, Marumo F, Sasaki S. 1997b.
Cloning and functional expression of a second
new aquaporin abundantly expressed testis.
Biochem Biophys Res Comm 237:714-718.
Jung JS, Preston GM, Smith
BL, Guggino WB, Agre P. 1994.
Molecular structure of the water channel through
Aquaporin CHIP. J Biol Chem
269:14648-14654.
Kashiwagi S, Kanamaru
K, Mizuno T. 1995. A
synechococcus gene encoding a putative
pore-forming intrinsic membrane protein. Biochim
Biophys Acta 1237:189-192.
King LS, Agre P.
1996. Pathophysiology of
the aquaporin water channels. Annu
Rev Physiol 58:619-648.
Kushmerick C, Rice SJ,
Baldo GJ, Haspel HC, Mathias RT. 1995.
Ion, water and neutral solute transport in Xenopus
oocytes expressing frog lens MIP. Exp
Eye Res 61:351-362.
Kyte J, Doolittle RF.
1982. A simple method for
displaying the hydropathy character of a protein.
J Mol Biol 157:105-132.
Le Cahérec F, Deschamps
S, Delamarche C, Pellerin I, Bonnec G, Guillam MT, Thomas
D, Gouranton J, Hubert JF. 1996.
Molecular cloning and characterization of an
insect aquaporin. Eur J Biochem
241:707-715.
Lee JW, Zhang Y, Weaver
CD, Shomer NH, Louis CF, Roberts DM. 1995.
Phosphorylation of nodulin 26 on serine 262
affects its voltage-sensitive channel activity in planar
lipid bilayers. J Biol Chem
270:27051-27057.
Livingstone CD, Barton
GJ. 1993. Protein
sequence alignments: A strategy for the hierarchical analysis
of residue conservation. Comput Applic
Biosc 9:745-756.
Luyten K, Albertyn J,
Skibbe WF, Prior BA, Ramos J, Thevelein JM, Hohmann S.
1995. Fps1, a yeast member
of the MIP family of channel proteins, is a facilitator
for glycerol uptake and efflux and is inactive under osmotic
stress. EMBO J 14:1360-1371.
Maurel C, Reizer J, Schroeder
JI, Chrispeels MJ. 1993. The
vacuolar membrane protein gamma-TIP creates water specific
channels in Xenopus oocytes.
EMBO J 12:2241-2247.
Maurel C, Tacnet F, Guclu
J, Guern J, Ripoche P. 1997.
Purified vesicles of tobacco cell vacuolar and
plasma membranes exhibit dramatically different water permeability
and water channel activity. Proc Natl
Acad Sci USA 94:7103-7108.
Modesto E, Lampe PD, Ribeiro
MC, Spray DC, Campos de Carvalho AC. 1996.
Properties of chicken lens MIP channels reconstituted
into planar lipid bilayers. J Membr
Biol 154:239-249.
Mulders SM, Knoers NVAM,
Van Lieburg AF, Monnens LAH, Leumann E, Wühl E, Schober
E, Rijss JPL, Van Os CH, Deen PMT. 1996.
New mutations in the AQP2 gene in nephrogenetic
diabetes insipidus resulting in functional but misrouted
water channels. J Am Soc Nephrol
8:242-248.
Mulders SM, Preston GM,
Deen PMT, Guggino WB, Van Os CH, Agre P. 1995.
Water channel properties of major intrinsic
protein of lens. J Biol Chem
270:9010-9016.
Pao GM, Wu LF, Johnson
KD, Höfte H, Chrispeels MJ, Sweet G, Sandal NN, Saier
MH Jr. 1991. Evolution
of the MIP family of integral membrane transport proteins.
Mol Microbiol 5:33-37.
Park JH, Saier HM Jr.
1996. Phylogenetic characterization
of the MIP family of transmembrane channel proteins.
J Membrane Biol 153:171-180.
Persson B, Argos P.
1994. Prediction of transmembrane
segments in proteins utilising multiple sequence alignments.
J Mol Biol 237:182-192.
Preston GM, Carrol TP,
Guggino WB, Agre P. 1992. Appearance
of water channels in Xenopus oocytes
expressing red cell CHIP28 water channel. Sciences
256:385-387.
Preston GM, Jung JS, Guggino
WB, Agre P. 1994. Membrane
topology of aquaporin CHIP. J Biol
Chem 269:1668-1673.
Rao JKM, Argos P.
1986. A conformational preference
parameter to predict helices in integral membrane proteins.
Biochim Biophys Acta 869:197-214.
Reizer J, Reizer A, Saier
MH Jr. 1993. The
MIP family of integral membrane channel proteins: Sequence
comparisons, evolutionary relationships, reconstructed
pathway of evolution, and proposed functional differentiation
of the two repeated halves of the proteins.
Crit Rev Biochem Mol 28:235-257.
Richey DP, Lin ECC.
1972. Importance of facilitated
diffusion for effective utilization of glycerol by Escherichia
coli. J Bacteriol
112:784-790.
Rivers RL, Dean RM, Chandy
G, Hall JE, Roberts DM, Zeidel ML. 1997.
Functional analysis of nodulin 26, an aquaporin
in soybean root nodule symbiosomes. J
Biol Chem 272:16256-16261.
Sabolic I, Brown D.
1994. Water transport in
renal tubules is mediated by aquaporins. Clin
Investig 72:698-700.
Saier MH Jr. 1994.
Computer-aided analysis of transport protein
sequences: Gleaning evidence concerning function, structure,
biogenesis and evolution. Microbio
Rev 58:71-93.
Sanders OI, Rensing S,
Kuroda M, Mitra B, Rosen BP. 1997.
Antimonite is accumulated by the glycerol facilitator
GlpF in Escherichia coli.
J Bacteriol 179:3365-3367.
Thompson JD, Higgins DG,
Gibson TJ. 1994. CLUSTAL
W: Improving the sensitivity of progressive multiple sequence
alignment through sequence weighting, positions-specific
gap penalties and weight matrix choice. Nucleic
Acids Res 22:4673-4680.
Weaver CD, Shomer NH,
Louis CF, Roberts DM. 1994.
Nodulin 26, a nodule-specific symbiosome membrane
protein from soybean, is an ion channel. J
Biol Chem 269:17858-17862.
Weig A, Deswarte C, Chrispeels
MJ. 1997. The major
intrinsic protein family of Arabidopsis
has 23 members that form three distinct groups with functional
aquaporins in each group. Plant Physiol
114:1347-1357.
Wistow G, Pisano MM, Chepelinsky
AB. 1991. Tandem
sequence repeats in transmembrane channel proteins.
Trends Biochem Sci 16:170-171.
Yang B, Verkman AS.
1997. Water and glycerol
permeabilities of aquaporins 1-5 and MIP determined
quantitatively by expression of epitope-tagged constructs
in Xenopus oocytes. J
Biol Chem 272:16140-16146.